Цирроз печени (ЦП) — это конечная стадия многих заболеваний печени, среди которых хронические гепатиты, алкогольная и неалкогольная жировая болезнь печени.1 Это процесс диффузного образования соединительной ткани, которая приводит к нарушению функции органа.
Цирроз печени необратим, медикаментозное и немедикаментозное лечение цирроза печени, как правило, направлено на предотвращение прогрессирования процесса фиброзирования и профилактику развития осложнений.
1 Распространенность хронических гепатитов и жировой болезни растет, особенно среди людей трудоспособного возраста — цирроз становится не только медицинской, но и социально и экономически значимой проблемой.
Распространенность заболевания на данный момент составляет более 20 миллионов.2 В РФ ежегодно регистрируется около 200 тысяч новых случаев цирроза печени ежегодно. 2
Причины цирроза
По статистике, около 50% цирроза печени связаны со злоупотреблением алкоголя и развитием алкогольной болезни печени (АБП). 1При этом у 25% из них отмечались вирусные гепатиты в анамнезе.1
Среди других причин развития цирроза выделяют2:
- болезни обмена, связанные с избыточным накоплением железа, меди и других элементов;
- Аутоиммунные поражения;
- Первичный и вторичный билиарный цирроз;
- Заболевания сосудистой системы (болезнь Бадда-Киари);
- Длительный прием лекарственных препаратов, которые оказывают токсическое действие на печень;
- неалкогольная жировая болезнь печени (НАЖБП), которая связана с накоплением большого количества свободных жирных кислот в гепатоцитах (печеночных клетках), характеризуется хроническим течением и потенциальным прогрессированием от стадии к стадии: стеатоз — стеатогепатит — фиброз — цирроз (так же, как и алкогольная болезнь печени, однако вероятность развития цирроза при АЖБП в 10 раз выше).5
Независимо от причины, развивается ЦП одинаково: нарушается дольковое строение печени с образованием узлов на месте разрушенных гепатоцитов— так называемых ложных долек.
- Мелкоузелковый — размеры фиброзных узлов не более 3 мм;
- Крупноузловой — узлы 3-5 мм;
- Cмешанный.
Симптомы цирроза
Клиника ЦП может быть очень разной — от полного отсутствия симптомов до появления признаков печеночной недостаточности, когда полностью нарушается работа печени1.
Чаще всего пациентов беспокоят:
- Сниженная трудоспособность; дискомфорт в животе,
- преимущественно в правом подреберье
Общая слабость и повышенная утомляемость
Диспепсия (симптомы несварения)
В дальнейшем появляются и другие симптомы, диагностику которых осуществляет исключительно врач.
Могут также наблюдаться:
- Зуд и пожелтение кожи
- Нарушения менструального цикла у женщин, импотенция у мужчин.
В дальнейшем появляются и другие симптомы, диагностику которых осуществляет врач
Диагностика
Диагностика цирроза печени начинается с клинического обследования, которое включает сбор жалоб и осмотр.
При осмотре врач может обнаружить внешние признаки ЦП1:
- Покраснение на ладонях и стопах (пальмарная эритема)
- Сосудистые звездочки
- Уменьшение волосяного покрова в подмышечных впадинах
- Белые ногти
- У мужчин с циррозом печени может отмечаться гинекомастия — увеличение грудных желез. Причинами этого считаются сниженный уровень тестостерона и повышенный уровень эстрадиола (женского полового гормона) в крови за счет различных механизмов: усиления процессов ароматизации тестостерона в эстрадиол, а также повышения продукции глобулина, связывающего половые стероиды, и снижения, таким образом, уровня свободного тестостерона.6,7
Позже возможно появление отеков, особенно на нижних конечностях, желтухи, развитие асцита — скопления свободной жидкости в брюшной полости.
При пальпации живота обнаруживается увеличение печени, нередко можно пропальпировать узловую поверхность органа, заостренный край. Однако в В конечной стадии болезни размеры печени могут уменьшиться, а селезенки — увеличиться.1 Для подтверждения диагноза и установления стадии заболевания проводятся лабораторные, инструментальные исследования, также используются расчетные индексы.
Лабораторные методы
- Стандартные биохимические анализы, которые определяют уровень билирубина, общего белка, протромбиновый индекс, сывороточное железо, ферритин и активность ферментов (АСТ, АЛТ, щелочная фосфатаза). Данные тесты неспецифичны, их отклонения могут наблюдаться и при других заболеваниях, но они помогают оценить степень нарушения функций печени.
- Общеклинические анализы крови могут выявить наличие анемии, снижения количества лейкоцитов, тромбоцитов, что может наблюдаться в том числе при ЦП.
- Тесты на вирусы гепатитов и возбудители других инфекций могут назначаться для определения потенциальной причины заболевания.
Расчетные индексы:
Использование тест-панелей, например, таких, как FibroTest, FibroIndex, Hepascore, может применяться для оценки стадии фиброза. Например, применение шкалы Fibrotest позволило избежать биопсии у 50% пациентов.2
Для оценки степени тяжести ЦП и прогноза выживаемости чаще всего используют классификацию Чайлд-Пью.2 Шкала, в которой учитываются результаты лабораторных тестов и клинические проявления, позволяет оценить степень функциональных нарушений и определить стадию болезни. Чем больше баллов, тем хуже прогноз.2
Инструментальные исследования
Кроме пункционной биопсии, которая признана «золотым стандартом» для диагностики фиброза и цирроза, важную роль в диагностике играют методы визуализации печени. К ним относятся:
- УЗИ печени, органов брюшной полости и забрюшинного пространства. Наиболее доступный и достаточно информативный метод. Позволяет оценить анатомические и структурные особенности, форму, размеры, сосудистый рисунок. Судить о ЦП можно по увеличению размеров печени и по ее неоднородной структуре и неровному бугристому контуру. Также при циррозе печени на УЗИ могут отмечаться признаки портальной гипертензии: асцит, расширение вен воротной системы, увеличение селезенки.1
- КТ, МРТ (компьютерная и магнитно-резонансная томография печени) — за счет выполнения большого количества тонких срезов дает полноценную информацию о структуре органа, наличии фиброзных узлов, ложных долек, дополнительных сосудов и т.д.
- Ультразвуковая эластометрия (аппарат FibroScan) может служить альтернативой биопсии тканей печени, так как дает возможность оценить плотность органа, возрастающую по мере увеличения процента соединительной ткани.2
Полную картину заболевания может дать только комплексная диагностика, поэтому используется комбинация различных методов, которые иногда приходится повторять неоднократно.
Осложнения цирроза печени
Осложнения при ЦП могут представлять угрозу жизни пациента, потому их своевременная диагностика — одна из первоочередных задач врача.
Асцит
Наиболее частое осложнение — в течение 10 лет формируется почти у 60% больных3. Его основные причины: повышение давления в системе воротной вены (портальная гипертензия) и недостаточное выделение натрия с мочой, что приводит к задержке жидкости.
Развитие у пациента с циррозом асцита ассоцируется с неблагоприятным прогнозом и снижением качества жизни пациентов.
Различают 3 степени тяжести этого состояния: от незначительного, который выявляется только при УЗИ, до напряженного, при котором асцит заметен невооруженным глазом3 (живот сильно увеличивается в размерах, сосуды расширяются, кожа натягивается, становится сухой, шелушится).
Асцит может спровоцировать развитие других осложнений.
Спонтанный бактериальный перитонит
При асците может произойти инфицирование жидкости в брюшной полости, причем источник и путь заражения обычно установить не удается. Дополнительные факторы — дефекты в иммунной системе и проникновение микроорганизмов из кишечника в асцитическую жидкость.
Симптомы перитонита: резкие боли в животе, напряжение передней мышечной стенки, общие признаки воспаления, шок, почечная недостаточность, желудочно-кишечное кровотечение. Однако у каждого пятого пациента он может протекать бессимптомно. Прогноз неблагоприятный.3
Гепаторенальный синдром
Гепаторенальный синдром (ГРС) характеризуется развитием почечной недостаточности на фоне тяжелых стадий ЦП.
Гепаторенальный синдром является диагнозом исключения, то есть устанавливается после исключения других заболеваний, которые могли стать причиной поражения почек.
Различают 2 типа гепаторенального синдрома — быстропрогрессирующий, который обычно связан с тяжелыми алкогольными гепатитами или развитием спонтанного бактериального перитонита, и медленнопрогрессирующий. Прогноз неблагоприятный при развитии любого типа ГРС3.
Печеночная энцефалопатия
Это целый комплекс психо-неврологических нарушений, которые в случае, если удается устранить причину, могут быть обратимы. У 80% больных с ЦП отмечаются признаки энцефалопатии3.
Симптомы: потеря внимания, замедление реакций, что особенно опасно для тех, кто управляет транспортными средствами. Лечение ПЭ эффективно, если начать его своевременно, то есть на стадии минимальной печеночной энцефалопатии.
По классификации West Haven различают 4 стадии: от рассеянности и невозможности сосредоточиться до выраженной дезориентации и комы.3
Портальная гипертензия и кровотечения из расширенных вен пищевода и желудка
Механизм развития осложнения: постоянное повышение давления в системе воротной вены приводит к варикозному расширению вен пищевода и желудка с кровотечением или без и асциту.
Диагностируется у половины пациентов с ЦП. Чем выше степень тяжести ЦП, тем больше риск развития портальной гипертензии. Наибольшую угрозу представляют кровотечения, так как они становятся причиной гибели 15-20% больных.
Портальная гипертензия может быть:
- Надпеченочной,
- Внутрипеченочной,
- Подпеченочной
От ее вида зависят подходы к лечению. При обнаружении гипертензии всем пациентам проводят профилактику кровотечений.3
Лечение цирроза
Лечение цирроза предусматривает:
- Лечение основного заболевания, которое привело к развитию цирроза;
- Специфическую терапия осложнений, в том числе медикаментозная: например, назначение диуретических препаратов при асците, назначение антибиотиков при спонтанном бактериальном перитоните и т.д.
- Хирургическое лечение асцита и кровотечений из расширенных вен пищевода и желудка1
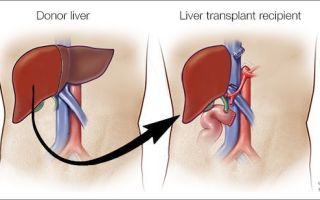
Важными составляющими являются диета, исключение вредных воздействий на печень, таких как алкоголь, гепатотоксические лекарственные препараты, лечение сопутствующих хронических болезней.4 В случае неэффективности принятых мер показана трансплантация органа1.
Публикации в СМИ
Гликогенозы — группа наследственных заболеваний, вызванных недостаточностью одного или нескольких ферментов, вовлечённых в синтез и распад гликогена, и характеризующихся накоплением патологических количеств или типов гликогена в тканях. Симптоматика возникает вследствие накопления гликогена или его промежуточных метаболитов или из-за недостатка конечных продуктов распада гликогена, особенно глюкозы. Гликоген и некоторые из промежуточных метаболитов, депонированных в тканях, могут быть обнаружены при МРТ. Различия в степени тяжести и возрасте начала клинических проявлений вызваны вовлечением различных изоферментов или других компонентов повреждённых ферментных систем. Частота всех форм болезней накопления гликогена — 1:40 000 населения.
Генетическая классификация и клиническая картина
• Гликогеноз типа 0 (*240600, 12p12.2, ген GYS2 [138571], r) — недостаточность гликоген синтетазы (КФ 2.4.1.11) печени. Клиническая картина: гипогликемия и гиперкетонемия натощак, судороги, гипергликемия и гиперлактатемия после приёма пищи.
• Гликогеноз типа I(a) (232200, 17q21, Â) — недостаточность глюкозо-6-фосфатазы (КФ 3.1.3.9), приводящая к избыточному накоплению гликогена нормальной химической структуры (особенно в печени и почках). Наблюдают значительно чаще других гликогенозов.
Клиническая картина: гипогликемия, артериальная гипертензия, задержка роста, позднее половое созревание, увеличение живота, аденомы печени, печёночноклеточная карцинома, гепатобластома, увеличение печени, хронический панкреатит, ксантомы, паукообразные гемангиомы, подагрические тофусы, протеинурия, гематурия, почечная недостаточность, центральный сегментарный гломерулосклероз, мочекислые камни почек, подагрический артрит, геморрагический диатез, лёгочная артериальная гипертензия. Лабораторные данные: недостаточность глюкозо-6-фосфатазы, гиперлипидемия, гиперурикемия, гиперлактацидемия, кетонемия, метаболический ацидоз. Синонимы: фон Гирке болезнь, нефромегалический гликогеноз, фон Гирке ван Кревельда синдром, фон Гирке ван Кревельда болезнь.
• Гликогеноз типа Ib (232220, r) — мутации гена транспортёра глюкозо 6 фосфата (*602671, 11q23.3). Клиническая картина: диарея, плохой аппетит, болезнь Крона, хронические остеомиелиты, перианальные абсцессы, нейтропения, гипохромная анемия, тромбоцитопения, вторичный амилоидоз, протеинурия, гиперлипидемия.
• Гликогеноз типа Ic (232240, r) — дефект транспортёра глюкозо 6 фосфата (*602671, 11q23.3).
Клиническая картина: гипогликемия, артериальная гипертензия, протеинурия, гематурия, почечная недостаточность, центральный гломерулосклероз, задержка роста и полового созревания, опухоли печени (аденомы, печёночноклеточная карцинома, гепатобластома), увеличение печени, хронический панкреатит, ксантома, ангиома кожи, подагрические тофусы, подагрический артрит, нормальные функции лейкоцитов, лёгочная артериальная гипертензия. Лабораторные данные: дефицит фосфат пирофосфат транслоказы, гиперлипидемия, гиперурикемия, гиперлактацидемия, кетонемия, метаболический ацидоз. ЛС (аллопуринол) следует назначать с осторожностью. Течение обычно хроническое прогрессирующее.
• Гликогеноз типа IIa (153360 — недостаточность лизосомной a-1,4 глюкозидазы, приводящий к избыточному накоплению гликогена нормальной химической структуры в сердце, скелетных мышцах, печени, мозге.
Клиническая картина: кардиомиопатия, кардиомегалия, артериальная гипотензия, миотония, мышечная слабость, утомляемость, увеличенный язык, смерть на первом году жизни, дыхательная недостаточность, одышка, аневризмы мозговых артерий. Синоним: Помпе болезнь.
• Гликогеноз типа IIb (300257, дефект ассоциированного с лизосомами мембранного белка LAMP2, r).
Клиническая картина: слабость проксимальных мышц, гипертрофическая кардиомиопатия, сердечная недостаточность, АВ-блокада, умственная отсталость.
Лабораторные данные: накопление гликогена в лизосомах; активность кислой мальтазы нормальная. Синонимы: болезнь Антополя, болезнь Данона (300257, дефект лизосомного белка LAMP2).
• Гликогеноз типа III (*232400, 1p21, ген AGL, GDE, r) — недостаточность амило-1,6-глюкозидазы, приводящая к накоплению гликогена ненормальной структуры с короткими внешними цепями в печени и мышцах.
Клиническая картина: миопатия, увеличение печени, гипогликемия, кетоацидоз, мышечная слабость с атрофией мышц, «ангельское» лицо, склонность к кровотечениям (в т.ч. носовым), гипертрофия желудочков на ЭКГ.
Синонимы: Кори болезнь, Форбса болезнь, лимитдекстриноз.
• Гликогеноз типа IV (*232500, 3p12, ген GBE1, r) — недостаточность 1,4-a-глюкан ветвящего фермента (КФ 2.4.1.18), приводящая к накоплению гликогена ненормальной структуры с длинными цепями в печени, почках, мышцах и других тканях.
Клиническая картина: цирроз печени, портальная гипертензия, печёночная недостаточность, гипогликемия, кардиомиопатия, сердечная недостаточность, миопатия, тазово-плечевая мышечная дистрофия, гиперлордоз позвоночника, походка вразвалку, слабость проксимальных мышц конечностей, смерть до 4-летнего возраста. Синонимы: болезнь Андерсен, амилопектиноз.
• Гликогеноз типа V (*232600, 11q13, ген PYGM, r) — дефект амилофосфорилазы (КФ 2.4.1.1), вызывающий накопление гликогена нормальной химической структуры в мышцах. Клиническая картина: слабость и атрофия скелетных мышц, мышечные боли при нагрузке, миоглобинурия. Синонимы: МакАрдла–Шмида–Пирсона болезнь, миофосфорилазная недостаточность, МакАрдла болезнь.
• Гликогеноз типа VI (*232700, 14q21–q22, ген PYGL, r) — недостаточность амилофосфорилазы (КФ 2.4.1.1), приводящая к накоплению гликогена нормальной структуры в гепатоцитах и лейкоцитах. Клиническая картина: увеличение печени, кетоз, гипогликемия, задержка роста. Синонимы: Гирса болезнь, гепатофосфорилазная недостаточность.
• Гликогеноз типа VII (*232800, 12q13.3, ген PFKM, r) — миопатии и увеличение печени, обусловленные недостаточностью 6 фосфофруктокиназы (КФ 2.7.1.11).
Клиническая картина: миопатия, увеличение печени, слабость мышц, крампи, гемолиз, лёгкая полицитемия, ретикулоцитоз, умеренная желтуха, желчнокаменная болезнь.
Лабораторные данные: недостаточность фосфофруктокиназы мышц, миоглобинурия, гиперурикемия. Синонимы: гепатофосфоглюкомутазная недостаточность, Томсона болезнь.
• Гликогеноз типа VIII (*261750, Xp22.2 p22.1, ген PHKA2, PHK; *311870, Xq12 q13, ген PHKA1, À) — недостаточность киназы фосфорилазы (КФ 2.7.1.38) в мышцах. Клинические и биохимические расстройства исчезают с возрастом, у большинства взрослых пациентов заболевание протекает бессимптомно.
Клиническая картина: увеличение печени, задержка роста, почечный канальцевый ацидоз.
Лабораторные данные: недостаточность печёночной киназы фосфорилазы (PHK); мышечная киназа фосфорилазы в пределах нормы; повышенное содержание глутамат-пируват и глутамат оксало-ацетат трансаминаз; гиперхолестеринемия; гипертриглицеридемия; кетонемия на фоне голодания; гиперлактацидемия или гиперурикемия отсутствует. Синонимы: Таруи болезнь, недостаточность киназы фосфорилазы печени и мышц. Примечание: некоторые субъединицы фермента имеют локусы в 16p12.1 p11.2.
• Гликогеноз типа VIIIb (261740, r) — крайне редкая форма недостаточности фосфофруктокиназы (КФ 2.7.1.38), ограниченная мышцей сердца.
• Гликогеноз типа VIIIc (*261750, r) — недостаточность киназы фосфорилазы (КФ 2.7.1.38) в печени и мышцах. Клиническая картина: увеличение печени, диарея, задержка роста, гипотония мышц, умеренная слабость. Лабораторные данные: недостаточность киназы фосфорилазы в печени и мышцах с накоплением гликогена.
ЛЕЧЕНИЕ • При типах 0, I и III — предотвращение гипогликемии и молочного ацидоза назначением дробных доз углеводов, что позволяет поддержать нормальные уровни глюкозы крови, предупредить развитие молочного ацидоза, гиперурикемии и гиперлипидемии.
Кроме того, используют непрерывную подачу высокомолекулярных декстранов через эндоназальный зонд.
Аллопуринол назначают для профилактики подагры и уратных камней почек • Ограничение анаэробной нагрузки уменьшает мышечные симптомы типов V и VII • При типе VIII рекомендуют ограничение физической нагрузки и обильное питьё • Эффективных методов лечения других типов нет.
МКБ-10 • E74.0 Болезни накопления гликогена
Цирроз печени — симптомы и лечение
Лечебные мероприятия начинаются с рациональной диеты. Она должна быть высококалорийной и высокобелковой (за исключением тяжёлых форм печёночной энцефалопатии), при асците — низкосолевой, с потреблением белка в определённом количестве (из расчёта на 1,5 г/кг массы тела и до 40 ккал/кг в день).
В качестве нутритивной поддержки рекомендуется дополнительные приёмы пищи в виде энтерального питания смесями, обогащёнными пищевыми волокнами, с низким содержанием ароматических аминокислот.
Этиотропное лечение
Этиотропная терапия предполагает лечение основного заболевания, на фоне которого развился цирроз:
- при вирусных гепатитах — противовирусная терапия аналогами нуклеозидов;
- при алкогольном стеатогепатите — исключение приёма алкоголя;
- при лекарственных стеатогепатитах в результате одновременного назначения множества лекарств — ограничение гепатотоксичных и нефротоксичных лекарственных препаратов с сохранением лекарства только по жизненным показаниям;
- при гемохроматозе — отмена препаратов железа.
Патогенетическая терапия
Лечение проводится в зависимости от механизмов развития ЦП, лечения обострений и наличия у пациента хронических заболеваний.
У пациентов с алкогольным циррозом печени (АЦП) возникает дефицит витаминов группы В. Поэтому в этих случаях необходимо принимать 100 мг тиамина, 30 мг пиридоксина и 1 мг фолиевой кислоты в сутки.
Для восстановления нарушенной структуры мембран гепатитов используют эссенциальные фосфолипиды, разведённые с кровью пациента по 10 мл на 500 мг в сутки в течение не более 14 дней.
Также можно использовать препараты селимарина («Силибинин», «Легалон») и адеметионина («Гептор», «Гептрал») по 800 мг в сутки в/в струйно, метионин и липовую кислоту.
При стойком холестазе и нехватке жирорастворимых витаминов в отсутствии признаков острой печёночной недостаточности в/в водят ретинол (витамин А), эргокальциферол (витамин D), токоферол (витамин Е), викасол (витамин К) и препараты кальция. Для купирования холестаза, развившегося на фоне АЦП, часто назначают препараты урсодезоксихолевой кислоты («Урсосан», «Урсофальк», «Урдокса», «Урсо 100»).
При аутоиммунном гепатите (АИГ) показана иммуносупрессивная и противовоспалительная терапия глюкокортикостероидами (преднизолон, метилпреднизолон). Приём азатиоприна позволяет снизить дозировку глюкокортикостеройдов. Вместо азатиоприна могут быть использованы 6-меркаптопурин, циклоспорин А, циклофосфамид, микофенолата мофетил.
| Преднизолон:60 мг — 1-я неделя;40 мг — 2-я неделя;30 мг — 3-я и 4-я неделя;20 мг — поддерживающая дозировка. | Преднизолон:30 мг — 1-я неделя;20 мг — 2-я неделя;15 мг — 3-я и 4-я неделя;10 мг — поддерживающая дозировка.Азатиоприн:50 мг — постоянно. |
Ориентировочный курс лечения — 22 месяца. Перед планируемой отменой терапии обязательно проводится контрольная биопсия печени, а после отмены — наблюдение не реже одного раза в шесть месяцев.
При первичном билиарном циррозе используют иммуносупрессивную терапию бедсонидом в сочетании с препаратами урсодезоксихолевой кислоты (УДХК), проводят мониторинг на предмет остеопороза. С появлением топических стеройдов с ММХ-технологией количество побочных действий от гормональной терапии снизилось.
При первичном склерозирующем холангите (ПСХ) длительно назначаются препараты УДХК в дозах 12-35 мг на 1 кг массы тела. Также возможно хирургическое лечение — эндоскопическая баллонная дилатация наиболее суженных участков желчных протоков. Данный вид лечения достоверно увеличивает продолжительность жизни пациентов с ПСХ
При гемохроматозе необходимо ограничить употребление железосодержащих продуктов и лекарственных препаратов.
Для коррекции уровня ферритина проводят лечебное кровопускание при помощи флеботомии до двух раз в неделю с периодической оценкой содержания гемоглобина и гематокрита в крови (до достижения ферритина сыворотки 50 мкг/л).
В случае противопоказаний к кровопусканию в связи с развитием анемии используют десферал (дефероксамин).
При болезни Вильсона — Коновалова показано ограниченное употребление продуктов, содержащих медь, и пожизненный приём хелатирующих соединений Д-пеницилламина (купренил, металкаптазу), триентина (куприд) или хелатирующих соединений цинка. При тяжёлом течении и терминальной стадии цирроза производят трансплантацию органа.
При недостаточности альфа-1-антитрипсина применяется только специфическая патогенетическая терапия с применением лечебных концентратов альфа-1-антитрипсина из донорской плазмы (Prolastin, Zemaira, Aralast).
При синдроме Бадда — Киари помимо ограничительной диеты производят коррекцию портальной гипертензии неселективными бета-адреноблокаторами (атенолон, пропранолол) и мочегонными препаратами (фуросемеид, спиронолактон).
Также используют антикоагулянтную терапию гепарином или варфарином до достижения целевого уровня МНО. При развитии эмболии проводится тромболитическая терапия стрептокиназой, урокиназой или рекомбинантным тканевым активатором плазминогена.
Основными методами лечения при синдроме Бадда — Киари являются:
- наложение шунтов для восстановления венозного кровотока;
- трансъюгулярное внутрипечёночное портосистемное шунтирование;
- ангиопластика при стенозе нижней полой вены.
При венокклюзионной болезни и обструкции воротной вены необходимо исключение этиологического фактора и антикоагулянтная терапия. При эмболии — тромболитическая терапия, коррекция портальной гипертензии и хирургическое лечение для наложения шунтов (тромбэктомия с ангиопластикой).
Симптоматическое лечение
Часто на фоне течения и декомпенсации ЦП развивается стойкий распространённый зуд. Для его купирования используют холестирамин, энтеросорбены и гемосорбенты (активированный уголь, энтеросгель, полисорб). Положительный эффект оказывает плазмаферез.
Для улучшения метаболических процессов восполняют витаминную недостаточность при помощи добавления препаратов УДХК. Некоторые пациенты с первичным билиарный циррозом эти препараты принимают пожизненно.
Хирургическое лечение
При неэффективности консервативного лечения, сохранённых резервах организма и учёте возрастных показателей принимают решение о трансплантации печени. Особенно это необходимо при выраженной гипоальбуминемии (менее 25 г/л) и стойкой гипербилирубинемии (более 100 мкмоль/л).
Лечение осложнений ЦП
Основная стратегия лечения осложнений — определение преобладающего компонента развития большой печёночной недостаточности и других осложнений.
Для лечения спонтанного бактериального перитонита назначается инвазионная антибактериальная терапия с учётом чувствительности микроба к антибиотикам (до получения результатов посева). Звенья оксидантного стресса блокируются при помощи диализных аппаратов MARS и Прометей.
Асцит лечится комплексно:
- обязательно ежедневное взвешивание и изменение окружности живота, контроль выпитой и выделенной жидкости (оптимальная скорость потери массы тела при лечении асцита — 0,5 л/сут);
- избегание назначения НПВП, строгая абстиненция;
- ограничение жидкости по диурезу (не требуется, если натрий сыворотки менее 125 ммоль/л);
- употребление пищи без добавления соли, бессолевого хлеба, сухарей;
- диуретическая терапия, включающая сочетание антагонистов альдостерона и петлевых диуретиков.
При напряжённом асците и неэффективности консервативного лечения используют парацентез (прокол и ликвидация жидкости). Если эвакуировано более пяти литров асцитической жидкости, показано в/в ведение альбумина из расчёта 10 г на литр эвакуированной жидкости.
Замедление прогрессирования цирроза
С этой целью используются препараты, обладающие антифибротическим действием — колхицин, интерферон и некоторые гепатопротекторы. Они уменьшают связывание коллагена и ускоряют его разрушение.[15][16]
Лечение цирроза печени народными средствами
Лечение методами альтернативной медицины не имеет научно обоснованных доказательств. Применяя народные средства, пациент может вовремя не получить эффективную терапию. В результате чего разовьются опасные осложнения: асцит, перитонит, энцефалопатия и острая почечная недостаточность.
Генетические заболевания печени
Болезнь Вильсона-Коновалова (БВК) – наследственное аутосомно-рецессивное заболевание печени и центральной нервной системы, характеризующееся хроническим и неуклонно прогрессирующим течением, связанное с накоплением токсических концентраций меди в печени, головном мозге, роговице глаза, почках и других органах. Мировая распространенность БВК составляет от 1 на 5000 до 1 на 30 000. Частота носительства дефектной аллели равна 1:90. Среди представителей малых народностей БКВ встречается чаще. Так, у евреев-ашкенази ее частота в 30 раз превышает среднемировую. Известно, что БВК является причиной болезней печени у детей в 15-20% случаев. Среди лиц моложе 35 лет с хроническим активным гепатитом 5% составляют пациенты с БКВ. В настоящее время считается, что это заболевание встречается значительно чаще, чем оно диагностируется. Причиной возникновения БВК являются мутации гена ATP7B, который расположен на длинном плече 13-й хромосомы. Ген кодирует трансмембранный белок, необходимый для выведения свободной меди с желчью и встраивания меди в плазменный белок-переносчик церулоплазмин. При нарушении функции данного белка медь в избыточном количестве откладывается в органах и тканях, что приводит к их повреждению.
БВК характеризуется длительным латентным течением и многогранностью клинической картины, которая включает в себя, прежде всего печеночные и неврологические проявления, а также поражения других органов и систем. Так исследователю J.Walsh (1972) принадлежат слова: «Нет ни одного больного с болезнью Вильсона, похожего один на другого».
В связи с полиморфизмом течения БВК относится к числу трудно диагностируемых заболеваний.
Одним из клинических диагностических признаков БВК является кольцо Кайзера—Флейшера, которое, однако, отсутствует у 50%-62% пациентов с печеночной манифестацией заболевания, зато может присутствовать у больных первичным билиарным циррозом, первичным склерозирующим холангитом.
Основными клиническими проявлениями БВК являются поражение печени: острый или хронический гепатит, цирроз печение, неалкогольная жировая болезнь печение – или стеатогепатит, а также фульминантный гепатит, а также различные виды гиперкинезов, атаксия, дистония, аффективные и поведенческие нарушения, психотические реакции, изменение когнитивных функций.
Среди лабораторных критериев БВК также нет строго патогномоничных. Концентрация церулоплазмина у пациентов с БВК обычно снижена, но у 15% пациентов с печеночной манифестацией болезни уровень церулоплазмина остается в пределах нормальных значений, так как его синтез увеличивается при воспалении ткани печени.
Ложно отрицательные результаты по уровню церулоплазмина могут встречаться и среди женщин, принимающих эстрогены, беременных. Повышение содержания церулоплазмина в сыворотке крови встречается также при таких состояниях как, рассеянный склероз, подострый склерозирующий панэнцефалит, болезнь Гентингтона.
Концентрация церулоплазмина в крови может быть пониженной у гетерозиготных носителей БВК, при циррозе печени другой этиологии, при мальабсорбции, у детей в возрасте до 2 лет, при врожденной гипо- или ацерулоплазминемии, болезни Менкеса.
Характерным для пацинетов с БВК является повышение суточной экскреции меди с мочой (более 50 мкг/сут) и повышение концентрации меди в моче. Ложноотрицательные результаты могут регистрироваться у детей на ранних стадиях заболевания, при бессимптомном течении.
Суточная экскреция меди с мочой может повышаться при холестатических заболеваниях печени, состояниях, сопровождающихся гепатоцеллюлярным некрозом, контаминации медью извне.
Таким образом, в связи с недостоверностью клинических и лабораторных маркеров БВК, единственным методом подтверждения диагноза является выявление дефектов в гене ATP7B.
К настоящему времени известно более 800 различных мутаций в гене ATP7B. При БВК встречаются миссенс-мутации, делеции, инсерции, нонсенс, фреймшифт мутации и мутации сайтов сплайсинга. Однако распространенность их среди определенных этнических групп неодинакова.
Наиболее частой мутацией у пациентов с БВК в европейских популяциях является точковая мутация с.3207С>А в экзоне 14. Она приводит к замене аминокислоты гистидина в положении 1069 на глутаминовую кислоту (His1069Gln). Среди россиян также распространены следующие мутации: c.
2532del.A (ех.10), с. 1770ins.T (ex.5), c. 2304insC (ex.8), c. 3627_3630del.4 (ex.17), c. 3649_3654del.6 (ex.17), с. 3942 del.АТ (ex.17), с. 3947 del.C (ex.17), c. 3026_3028 del.TCA (ex.13), c. 3029insT (ex.13), c. 3031insC (ex.13), с. 1340_1343 del.4 (ex.3), с. 3402 del.
(ex. 15).
Наследственный гемохроматоз (НГ) представляет собой спектр наследственных заболеваний, возникающих в результате мутаций в генах, участвующих в метаболизме ионов железа.
Аберрации в данных генах ведут к чрезмерному накоплению железа в тканях и органах, что вызывает нарушение их функций из-за повышенной продукции свободных радикалов. В 85-90% случаев причиной НГ являются мутации в гене HFE (High Iron Fe).
Наиболее частая генетическая аберрация, приводящая к развитию НГ, является гомозиготная мутация C282Y в гене HFE, обнаруживаемая в 80-85% случаев. Кроме этого, гетерозиготная мутация C282Y, ассоциированная с гетерозиготными или гомозиготными полиморфизмами H63D и S65C, также приводит к развитию клинической симптоматики НГ.
Нужно отметить, что пенетрантность мутаций в гене HFE составляет 70% и только у 10% пациентов наблюдается тяжелое мультисистемное поражение органов. Распространенность мутации C282Y может достигать 1:200, в то время как распространённость тяжелых проявлений НГ достигает 1:2000.
Ранними симптомами НГ являются артралгия, хроническая усталость, импотенция у мужчин и аменорея у женщин, потеря волос, а также гепатомегалия, стеатогепатит, гиперпигментация кожных покровов и артрит. В дальнейшем у пациентов с НГ может развиваться тяжелое нарушение функций печени вплоть до цирроза, а также сахарный диабет 2 типа и кардиомиопатия.
Обнаружение гомозиготной мутации C282Y, а также компаундной гетерозиготы C282Y/ H63D и C282Y/ S65C в гене HFE с классической картиной НГ подтверждает клинический диагноз. При обнаружении перечисленных аберраций у бессимптомного носителя имеется высокий риск развития симптомов НГ в будущем.
Генетические аспекты формирования неалкогольной жировой болезни печени | #08/19 | «Лечащий врач» – профессиональное медицинское издание для врачей. Научные статьи
Неалкогольная жировая болезнь печени (НАЖБП) является одной из наиболее распространенных причин хронической болезни печени.
В настоящее время НАЖБП имеет предполагаемую распространенность в общей популяции от 20% до 30% в западных странах и от 5% до 18% в Азии и, по прогнозам, со временем возрастет [1–4].
Примечательно, что общая распространенность НАЖБП среди детей достигла примерно 10%, с тревожным показателем распространенности 17% у подростков [5]. Эта распространенность резко увеличивается от 40% до 70% у детей с ожирением [6–7].
От 3% до 5% пациентов с НАЖБП могут заболеть ранним неалкогольным стеатогепатитом (НАСГ) [8], характеризующимся долевыми и портальными воспалительными инфильтратами, происходящими из моноцитов, макрофагов, нейтрофилов и лимфоцитов; различной степенью фиброза, гибелью гепатоцитов и патологическим ангиогенезом [9].
Изотопные исследования биопсий печени у пациентов с ожирением, гипертриглицеридемией, гиперинсулинемией, имеющих НАЖБП, выявили повышенные уровни потока свободных жирных кислот, полученных из жировой ткани, и липогенез de novo [10], а также нарушение окисления и секреции жирных кислот [11].
Модулированная экспрессия и/или секреция транскрипционных факторов и цитокины, соответственно, влияют на последующие метаболические пути и, таким образом, играют решающую роль в патогенезе НАЖБП.
Например, регуляторный элемент, связывающий стерол-регуляторный белок 1с (SREBP-1c/SREBF1), контролирует экспрессию липогенных генов. Повышенный SREBP-1c коррелирует с печеночным стеатозом у пациентов с НАЖБП [12].
Отсутствие элемента, связывающего белок (ChREBP/MLXIPL), который регулирует метаболизм глюкозы и липогенез, облегчает течение стеатоза печени, что указывает на то, что ChREBP также связан с НАЖБП [13].
Помимо факторов транскрипции, участвующих в эндогенном липидном обмене, ядерные рецепторы, регулирующие метаболизм ксенобиотиков, такие как рецептор прегнана X (PXR/NR1I2), конститутивный андростановый рецептор (CAR/NR1I3) и ферменты метаболизма лекарств, также имеют измененные показатели у пациентов с НАЖБП [14].
Пататин-подобный фосфолипазный домен 3 (PNPLA3)
PNPLA3 является одним из немногих примеров, которые были подтверждены в нескольких популяциях и убедительно показали общую связь с НАЖБП. Экспрессия PNPLA3 регулируется ChREBP и SREBP, которые влияют на его функцию в метаболизме глюкозы и липидов [15].
Удивительно, но мыши с повышенной экспрессией PNPLA3 не показали нарушения липолиза или наличие стеатоза печени [16].
Вариант rs738409 [G] значительно ассоциируется с повышенным печеночным накоплением жировых клеток в печени и воспалением, тогда как rs6006460 [T] коррелирует с низким содержанием печеночного жира.
Это наблюдение было обнаружено в финском исследовании [17] и недавнем крупномасштабном исследовании геномных ассоциаций GWAS (GWAS — Genome-Wide Association Studies), в котором проводилось генотипирование 2,4 млн полиморфных единичных нуклеотидов у 7100 человек в качестве метаанализа из нескольких крупных популяционных исследований [18]. Кроме того, исследование населения Китая, Индии, Малайзии, Японии и Соединенного Королевства указало, что к наличию гомозиготности в варианте rs738409 [G] предрасположены пациенты с неалкогольным стеатогепатитом (НАСГ) [19–21]. G-аллель rs738409 PNPLA3 также был чрезмерно представлен при алкогольном/метаболическом циррозе [22].
Марганцевая супероксиддисмутаза (MnSOD/SOD2)
Так как окислительный стресс является одним из главных механизмов в развитии НАСГ, то молекулярные механизмы, которые приводят к генерации активных форм кислорода (АФК), могут способствовать патогенезу НАСГ. SOD2 защищает от АФК путем детоксикации супероксидов в кислород и перекись водорода.
T1183C был идентифицирован как ген, который направляет SOD2 в митохондрии [23]. У пациентов с НАЖБП наблюдалось снижение транспорта белка в митохондрии.
Эта связь была дополнительно подтверждена семейным анализом с использованием теста неравновесия передачи в 55 информативных семьях европейской популяции с НАЖБП [24].
Фактор некроза опухоли α (ФНО-α)
ФНО-α является цитокином с широким спектром функциональных возможностей при воспалении, иммунном ответе, апоптозе опухолевых клеток и метаболической регуляции организма [25].
Рост активности ФНО-α коррелирует с инсулинорезистентностью и степенью активности воспалительных процессов, таким образом увеличивая риск прогрессирования НАЖБП в стеатогепатит [26]. Было выявлено несколько различных вариантов промотора гена ФНО-α.
Варианты гена изучались на итальянской популяции, и была определена более высокая распространенность промотора ФНО-α-308 у контрольной группы, по сравнению с пациентами, имеющими НАЖБП, у которых более распространен вариант ФНО-α-238 [27].
Несмотря на нарушение чувствительности к инсулину, вариант ФНО-α-238 также был связан с пониженным уровнем холестерина, липопротеинов низкой плотности (ЛПНП) и сравнительно более низким индексом массы тела (ИМТ), что указывает на обратное влияние на регуляцию глюкозы и липидного обмена [28].
Фосфатидилэтаноламинметилтрансфераза (phosphatidylethanolamine N-methyltransferase, PEMT)
PEMT способствует синтезу фосфатидилхолина из фосфатидилэтаноламина. Фосфатидилхолин является важным компонентом формирования липопротеинов очень низкой плотности (ЛПОНП) для секреции триглицеридов (ТГ) печени. В экспериментальном исследовании было показано, что потеря функции PEMT приводит к увеличению накопления липидов у мышей [29].
Аполипопротеин Е (apolipoprotein E, ApoE)
ApoE является основным компонентом липопротеинов, и его роль в патогенезе НАЖБП широко признана.
Экспериментальные исследования, проведенные на грызунах, показывают пониженную восприимчивость к развитию ожирения и НАЖБП при отсутствии ApoE [30]. Ген ApoE имеет варианты ε2, ε3 и ε4.
Встречаемость аллеля ApoE ε3 значительно выше у представителей кавказских национальностей с НАСГ по сравнению с контрольной группой, в то время как наличие аллеля ApoE ε2 может защищать от НАЖБП [31].
Адипонектин (АН)
АН выполняет многочисленные системные защитные функции, такие как повышение чувствительности тканей к инсулину, противовоспалительные, антифиброгенные и антиатерогенные эффекты, участвует в церамидном катаболизме и подавлении глюконеогенеза в печени.
Генотипы полиморфизма (SNP, Single nucleotide polymorphism) G45T и G276T — наиболее часто встречающиеся варианты гена, кодирующего АН (ADIPOQ), в итальянской когорте пациентов с НАЖБП, по сравнению с контрольной группой [32].
АН в печени связывается с рецептором ADIPOR2, который отрицательно коррелирует с инсулинорезистентностью [33].
Вариант rs767870 гена, кодирующего рецептор адипонектина ADIPOR2, значительно чаще ассоциируется с печеночным накоплением липидов, наблюдаемым в финской когорте и подтвержденным в двух независимых исследованиях [34].
Рецепторы инсулина
Инсулинорезистентность является обычным явлением при НАЖБП и НАСГ.
Воспалительные факторы нацелены на субстрат рецепторов инсулина для осуществления убиквитин-опосредованной деградации белков через активацию супрессоров передачи сигналов цитокинов 3 (SOCS-3), вызывая тем самым подавление чувствительности к инсулину [35].
Больные с НАЖБП и сахарным диабетом (СД) 2 типа обычно лечатся PPARγ-таргетированным тиазолидиндионом, сенситайзером инсулина, который уменьшает высвобождение липидов, вызывает поглощение и хранение липидов и подавляет печеночный глюконеогенез [36].
Гормон роста (ГР) и инсулиноподобный фактор роста (ИФР)
НАЖБП и другие варианты метаболического синдрома часто встречаются у пациентов с синдромом дефицита гормона роста взрослых и гипопитуитаризмом. ГР оказывает активное влияние на окисление жирных кислот, липолиз, клиренс ЛПНП и глюконеогенез [37].
Большинство этих эффектов осуществляется через взаимодействие ГР с ИФР-1, который является катаболическим гормоном, секретируемым гепатоцитами при стимуляции ГР.
Исследования, проводимые на грызунах, также выявили ГР-независимые эффекты ИФР-1, заключающиеся в улучшении течения стеатогепатита путем подавления окислительного стресса [38].
Пациенты, имеющие ожирение, синдром дефицита гормона роста взрослых, НАЖБП и резистентность к инсулину, показывают низкий уровень ГР и ИФР-1 и увеличение ГР-связывающего белка, который коррелирует со стадиями стеатоза и фиброза печени [39]. ГР-заместительная терапия уменьшает окислительный стресс у пациентов с НАСГ, с синдромом дефицита гормона роста взрослых [40].
Циркадные ритмы — еще один фактор риска заболеваний печени
В печени циркадный ритм является результатом взаимодействия цис-регуляторных элементов [41]. Было выявлено несколько циркадных факторов транскрипции, которые могут включать и выключать экспрессию генов, но знания о глобальных изменениях ритмической транскрипции на уровне генома только начинают накапливаться [42].
Циркадные процессы на уровне тела включают цикл сна/бодрствования, температуру тела, кровяное давление, секрецию гормонов и т. д. Концентрация нескольких метаболитов крови, включая холестерин [43] и кортикостерон [44], аналог кортизола у мышей, изменяется в течение 24 часов.
Таким образом, неудивительно, что неповрежденные циркадные часы необходимы для поддержания гомеостаза организма [45] и что нарушение часов приводит к десинхронизации метаболизма и, следовательно, к патологиям, включая ожирение и рак [46]. Кроме того, часовые гены были определены как потенциальные терапевтические мишени.
Небольшая синтетическая молекула KL001 связывается с криптохромом CRY1 и предотвращает его деградацию, что приводит к удлинению циркадного периода. Стабилизация белка CRY имеет важные метаболические последствия, так как он ингибирует печеночный глюконеогенез [47].
Основными причинами нарушения работы часов являются хронические нарушения образа жизни, профессиональные факторы или длительная сменная работа. Экспериментальные исследования показали, что, по-видимому, люди и мыши имеют сходные механизмы, поскольку циркадная десинхрония также способствует нарушению обмена веществ в мышиной модели сменной работы [48].
Поскольку связь между печеночными часами и метаболическими нарушениями у людей трудно оценить, мышиные модели остаются важными инструментами [49]. Животные с мутациями в генах часов предоставили ключевую информацию о взаимозависимости между циркадными часами и метаболизмом [50].
Одно из самых ярких доказательств существенной связи между часами и метаболизмом у людей вытекает из 14-летнего проспективного исследования, проведенного в Японии, в котором около 7000 работников сталелитейной промышленности регулярно обследовались на предмет общего состояния здоровья и липидных показателей крови.
В ходе исследования было установлено неблагоприятное влияние чередующейся сменной работы на липидный обмен, что привело к статистически значимому повышению уровня общего холестерина в крови [51].
Последствия разрушения часов при заболеваниях кишечной системы, включая воздействие на печень, также были рассмотрены [52], но прямая связь между нарушением часов и заболеваниями печени была показана только на моделях мышей. Фиброз печени у мышей приводит к изменениям циркадного ритма и генов печеночных часов, где суточный ритм криптохрома CRY2 заметно снижается и связан с потерей цитохром-P450-редуктазы (POR) [53]. Более того, нарушение циркадного ритма ускоряет канцерогенез в печени у мышей, указывая на то, что постоянная циркадная координация может замедлять или возобновлять развитие рака [54].
Другое исследование сообщило о связи между общими генетическими вариациями циркадого гена CLOCK и восприимчивостью к НАЖБП [55]. В исследование были включены 136 пациентов с НАЖБП и 64 здоровых человека, и было показано, что частоты гаплотипов варианта CLOCK значительно различаются между пациентами с НАЖБП и контрольной группой.
Новые показания указывают на то, что ферменты модификации гистонов являются дополнительными мишенями генов, связанных с нарушениями циркадного ритма. Циркадная модификация гистоновых областей в ежедневно активируемых генах подчеркивает эпигенетическую модификацию как ключевой узел, регулируемый часами.
Было обнаружено, что гистон-ремоделирующий фермент MLL3 модулирует сотни эпигенетически нацеленных циркадных «выходных» генов печени. Таким образом, MLL3 является ферментом, связывающим циркадные часы и заболевания печени.
Можно полагать, что другие гены и метаболиты, связанные с циркадным ритмом, могут в будущем стать признанными многообещающими неинвазивными маркерами, которые будут полезны при определении стадий и прогноза заболеваний печени.
Заключение
Сложный патогенез НАЖБП отражает гетерогенную природу заболевания с точки зрения прогрессирования, серьезности и восприимчивости в разных этнических группах.
Хотя исследование GWAS и другие генные подходы выявили несколько факторов восприимчивости, связанных с НАЖБП, только немногие (в случае НАЖБП только PNPLA3) были подтверждены в нескольких популяциях.
Большинство исследований имеют недостаточную статистическую выборку, не имеют хорошо подобранных образцов и отсутствие различных стадий заболевания.
Биопсия печени остается золотым стандартом для диагностики НАСГ, но большинство современных исследований основано на ультразвуковой идентификации стадии заболевания, у которой не хватает точности из-за невозможности различить стеатоз от ранних стадий НАСГ. Дальнейшее развитие комбинированной оценки рисков полиморфизма в сочетании с секвенированием всего генома может помочь в выявлении групп высокого риска для профилактики и раннего выявления заболевания.
Литература
- Masarone M., Federico A., Abenavoli L. et al. Non alcoholic fatty liver: epidemiology and natural history // Rev Recent Clin Trials. 2014; 9: 126–133.
- Loomba R., Sanyal A. J. The global NAFLD epidemic // Nat Rev Gastroenterol Hepatol. 2013; 10: 686–690.
- Brunt E. M. Pathology of nonalcoholic fatty liver disease // Nat Rev Gastroenterol Hepatol. 2010; 7: 195–203.
- Adams L. A., Lymp J. F., St. Sauver J. et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study // Gastroenterology. 2005; 129: 113–121.
- Patton H. M., Sirlin C., Behling C. et al. Pediatric nonalcoholic fatty liver disease: a critical appraisal of current data and implications for future research // J Pediatr Gastroenterol Nutr. 2006; 43: 413–427.
- Schwimmer J. B., Newton K. P., Awai H. I. et al. Paediatric gastroenterology evaluation of overweight and obese children referred from primary care for suspected non-alcoholic fatty liver disease // Aliment Pharmacol Ther. 2013; 38: 1267–1277.
- Schwimmer J. B., Pardee P. E., Lavine J. E. et al. Cardiovascular risk factors and the metabolic syndrome in pediatric nonalcoholic fatty liver disease // Circulation. 2008; 118: 277–283.
- Williams C. D., Stengel J., Asike M. I. et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study // Gastroenterology. 2011; 140: 124–131.
- Vernon G., Baranova A., Younossi Z. M. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults // Aliment Pharmacol Ther. 2011; 34: 274–285.
- Donnelly K. L., Smith C. I., Schwarzenberg S. J. et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease // J. Clin. Invest. 2005; 115: 1343–1351.
- Kohjima M., Enjoji M., Higuchi N. et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease // Int. J. Mol. Med. 2007; 20: 351–358.
- Yang Z. X., Shen W., Sun H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease // Hepatol. Int. 2010; 4: 741–748.
- Dentin R., Benhamed F., Hainault I. et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice // Diabetes. 2006; 55: 2159–2170.
- Naik A., Beliс A., Zanger U. M. et al. Molecular interactions between NAFLD and xenobiotic metabolism // Front. Genet. 2013; 4: 2.
- Dubuquoy C., Robichon C., Lasnier F. et al. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes // J. Hepatol. 2011; 55: 145–153.
- Basantani M. K., Sitnick M. T., Cai L. et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome // J. Lipid Res. 2011; 52: 318–329.
- Kotronen A., Johansson L. E., Johansson L. M. et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans // Diabetologia. 2009; 52: 1056–1060.